


Main research activities
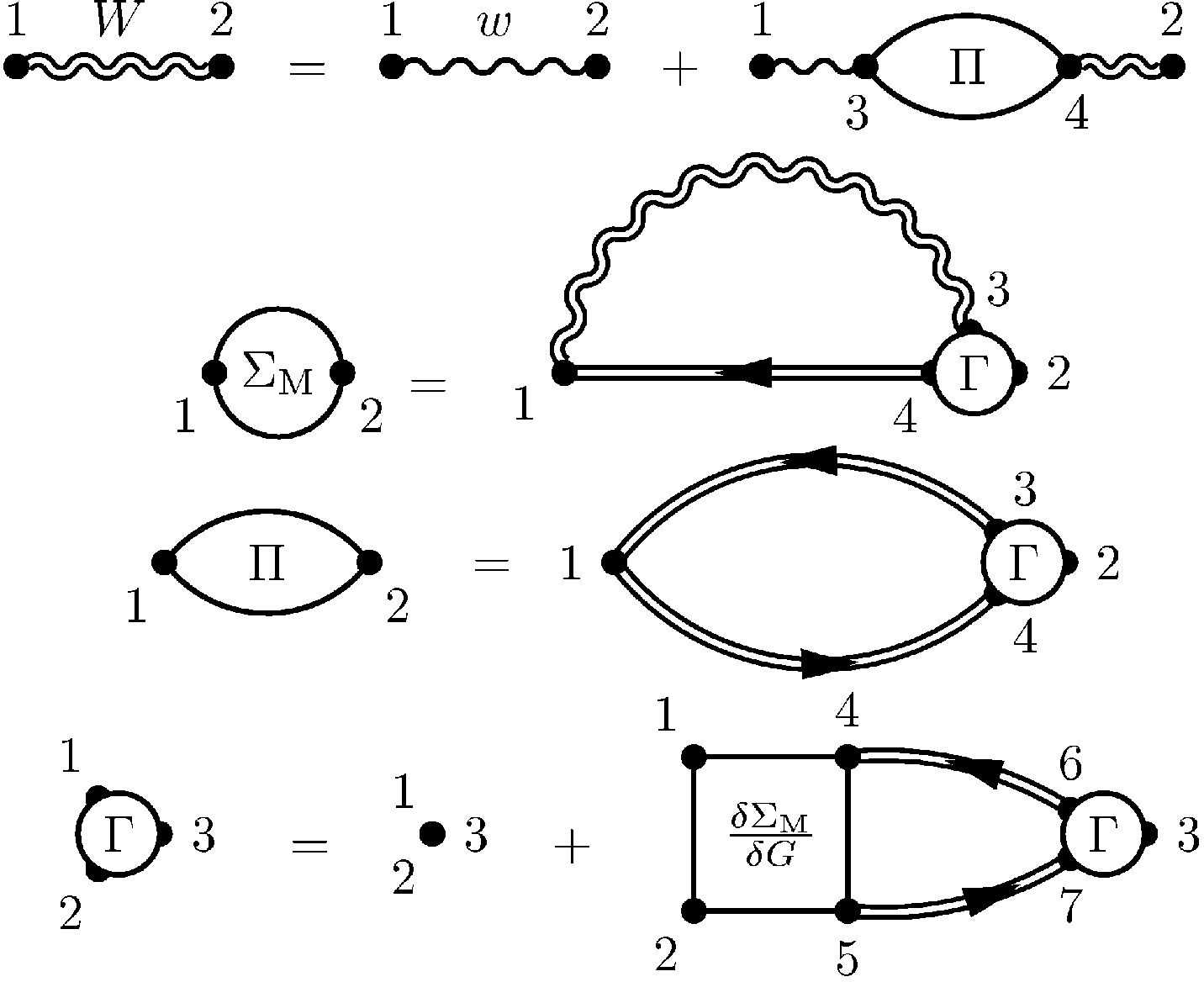
Ab initio many-body perturbation theory: the Fiesta initiative
In the present field of condensed-matter physics, many-body perturbation theory represent a family of techniques aiming at calculating
the correlation energy of electronic systems in general, including electon-electron, electron-hole, electron-phonon etc. interactions,
going beyond mean-field approaches such as Hartree-Fock or density functional (DFT) theories. A specific class of techniques developed
in the mid-60s relies on the inclusion of 2-body, 3-body etc. interactions, as in standard perturbation theory, but considering the
dynamically screened Coulomb interaction W(r,r'; ω) instead of the (stronger) bare Coulomb potential 1/|r-r'|. The GW and
Bethe-Salpeter formalisms are low-order approximation for describing electron-electron and electron-hole interactions within this family
of techniques. They have been shown to yield quasiparticle energies ("band structures") and optical (excitonic) spectra in much better
agreement with experiment as compared e.g. to standard DFT, for a very large class of systems.
In order to study organic systems with a good tradeoff between computer time and accuracy, and also to have a tool that we can develop
as a mean to implement and test new approximations and functionalities, we are developing a real-space Gaussian-basis
GW and Bethe-Salpeter code, the Fiesta package, allowing to study systems comprizing a few hundred atoms with "reasonnable" computer
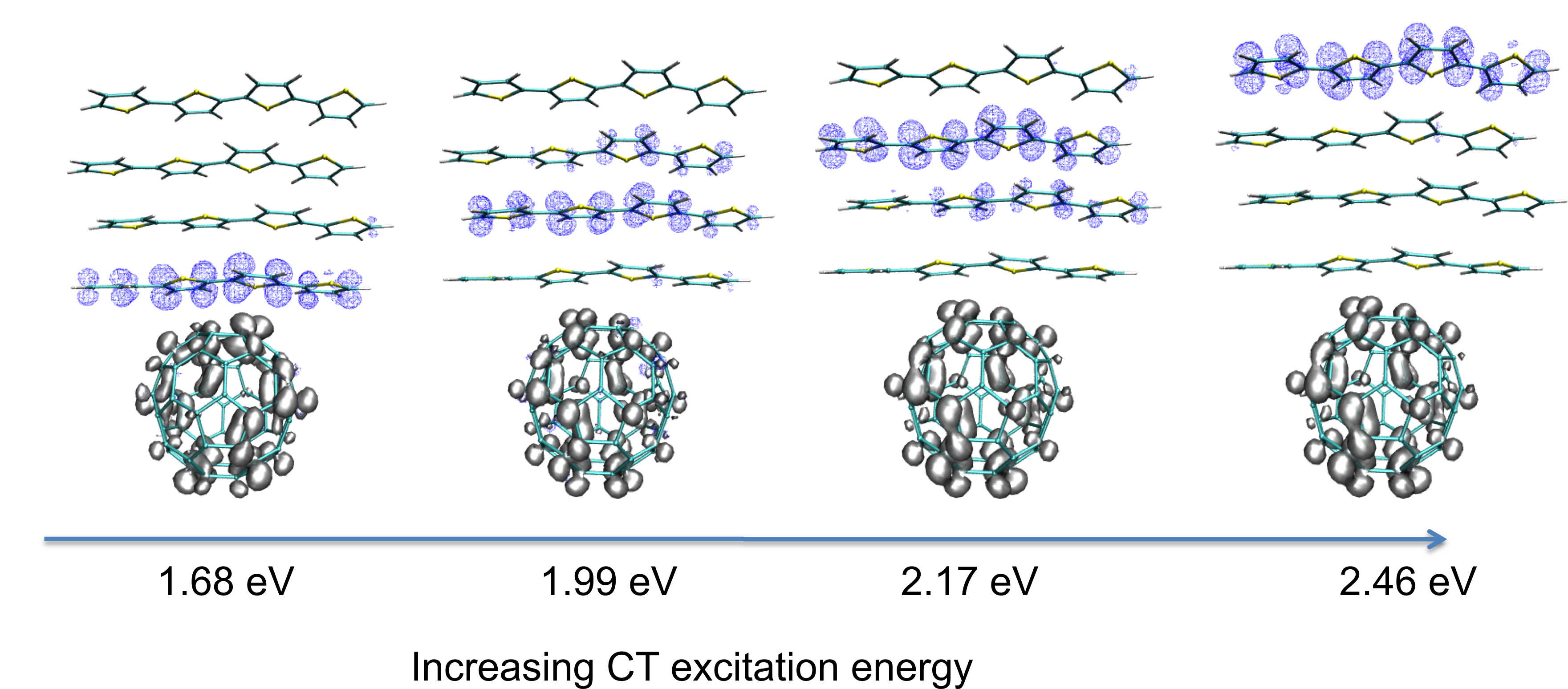
resources. A first class of applications, which motivated the Fiesta initative, concerns organic systems for photovoltaic applications.
Visit the Fiesta homepage.
Organic photovoltaics
Organic photovoltaics is concerned with photovoltaic systems (solar cells) of which the photoactive material is made of organic
molecules or small polymers, instead e.g. of silicon thin films. Even though providing today power conversion efficiencies still
smaller than standard inorganic cells, the low cost and large variety of active molecules or polymers offer much perspectives
for the years to come. Further, the basic mechanisms believed to contribute to the physics or chemistry of these systems, such as
charge/energy transfer excitations and electron-vibration (polaronic) coupling, are also at the heart of a large variety of
exciting fields, including photosynthesis or photochemistry. Despite their importance, such physical phenomena are still very
badly understood, as they rely on a complex interplay between (hot) electrons, holes, excitons and vibrational modes. Ab initio
simulations, in hands with model Hamiltonians, can act as a "theoretical microscope" to try to "observe" and understand the relevant
mechanisms, in order to propose new materials or new ideas. The study of such systems, composed of a disordered mixing of molecular
building blocks comprizing up to a hundred atoms each, is still a challenge to "accurate enough" quantum theories, so that both
methodology developments and applications to systems of increasing realism as compared to experiment, must be pursued at the same
time. This is in particular the origin of our Fiesta initiative above mentioned. Selected publications:
• First-principles GW calculations for fullerenes, porphyrins, phtalocyanine, and other molecules of interest for organic
photovoltaic applications, Blase et al. Phys. Rev. B 83, 115103 (2011).
• Electron-phonon coupling in the C60 fullerene within the many-body GW approach,
Faber et al. Phys. Rev. B 84, 155104 (2011).
• Short-range to long-range charge-transfer excitations in the zincbacteriochlorin-bacteriochlorin complex: A Bethe-Salpeter study,
Duchemin et al. Phys. Rev. Lett. 109, 167801 (2012).
• Fast and Accurate Electronic Excitations in Cyanines with the Many-Body Bethe-Salpeter Approach,
Boulanger et al. J. Chem. Theory Comput. 10, 1212-1218 (2014).
Superconductivity in doped semiconductors
When heavily doped, semiconductors or insulators undergo the so-called Mott insulating-metallic transition.
This subject was initiated in 2003 by the study of polymerized cage-like low-density sp3 silicon
phases (clathrates) allowing heavy intercalation doping (D. Connetable PhD thesis). The ab initio study
of the electron-phonon coupling matrix elements, combined with the Eliashberg formalism and the phonon-mediated
BCS picture, allows to provide insight into the superconducting mechanisms and the typical transition temperature
(Tc). Our discussion of the superconducting transition in silicon clathrates in 2003 was followed by the
discovery of a superconducting transition in heavily B-doped diamond (2004) and silicon (2006).
Selected publications:
• Superconducting Group IV Semiconductors, Blase et al. Nature Materials 8, 375-382 (2009)
• Superconductivity in doped cubic silicon, Bustarret et al. Nature 444, 465-468 (2006)
• Role of the Dopant in the Superconductivity of Diamond, Blase et al. PRL 93, 237004 (2004)
• Superconductivity in doped sp3 semiconductors: the clathrates,
Connetable et al. PRL 91, 247001 (2003).
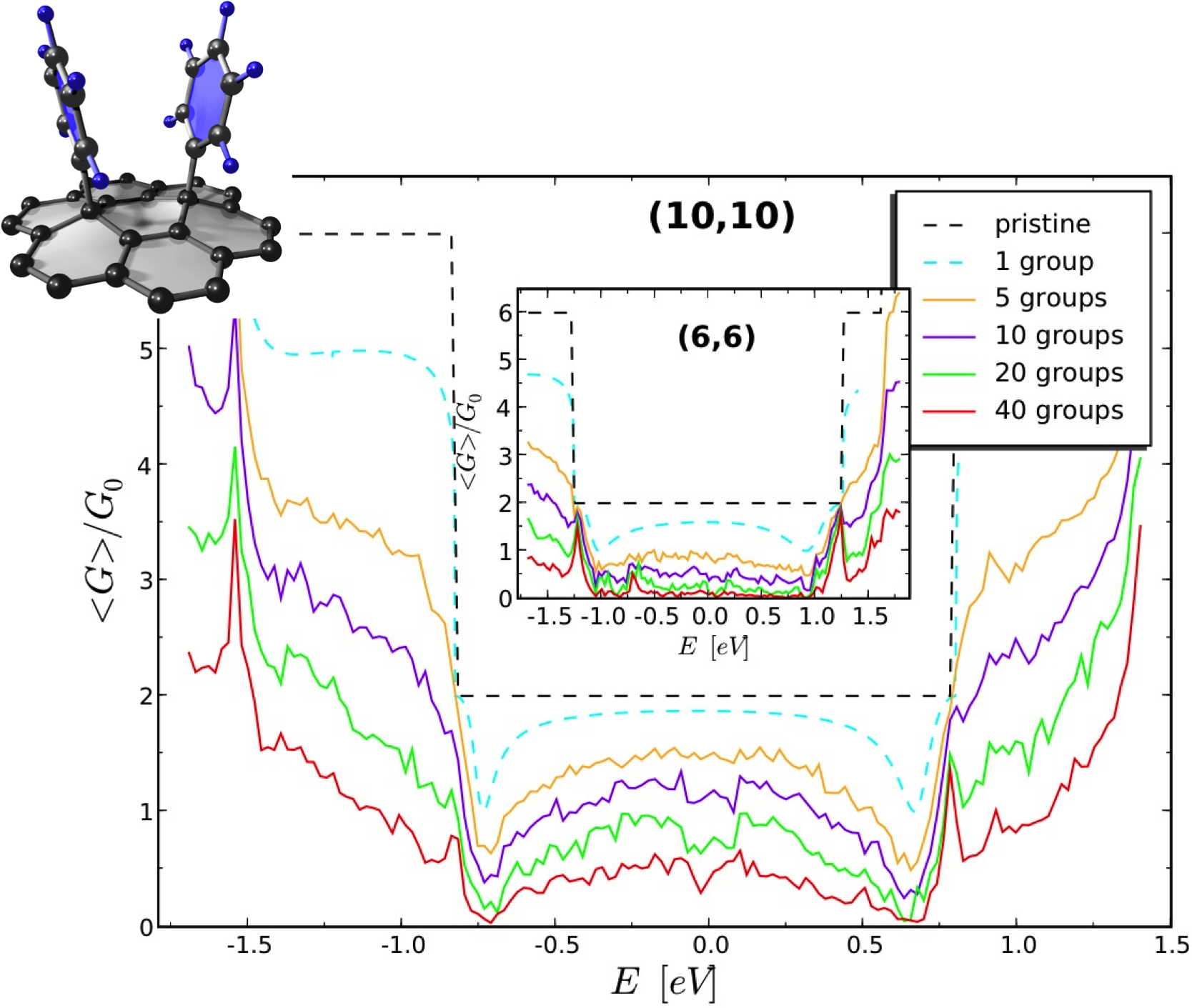
Electronic transport in nanotubes and graphene
Amongst all potential applications relying on the mechanical, electronic, or optical properties of
nanotubes and graphene, their electronic conductivity has been generating much work with early experimental
demonstrators of active transistors where the canal is a nanotube or graphene. Several difficult issues are
related to our ability to doped, functionalize, and in general taylor the properties of the conducting canal
without destroying the exceptional transport properties of the pristine tube, graphene ribbon, etc. Following
an early initiative to develop a simple ab initio code calculating the conductance of 1D systems within
a Green's function formalism restricted to the linear regime (code Tablier, Adessi/Blase), we have developed
a collaboration with Stephan Roche (CEA Grenoble, now in Barcelona) to bridge the gap between ab initio
simulations at the nanoscale, and mesoscopic physics concerned with the study of systems of which the size
is larger than the scattering length. We could calculate the conductance of micrometer long tubes, wires or
ribbons randomly doped or functionalized, averaging over hundreds of disorder configurations, to calculate
the average conductance as a function of incoming wavepacket energy, and the related scattering and localization
lenghts. While restricted to the linear regime, such ab initio studies could provide clear information
on the type of doping or functionalization that would optimally preserve as much as possible the conductance
of the charge-carrying canal. Selected publications:
• Effect of the Chemical Functionalization on Charge Transport in Carbon Nanotubes at the Mesoscopic Scale, Lopez-Bezanilla et al. Nano Lett. 9, 940944 (2009)
• Anomalous Doping Effects on Charge Transport in Graphene Nanoribbons, B. Biel et al. PRL 102, 096803 (2009)
• Charge Transport in Chemically Doped 2D Graphene, Lherbier et al. PRL 101, 036808 (2008)
• Electronic and transport properties of nanotubes, Charlier et al. RMP 79, 677 (2007)